Published On May 27, 2020
EVEN AS THE WORLD was learning about an unusual viral pneumonia surging in Wuhan, China, scientists at the National Institute of Allergy and Infectious Diseases (NIAID) were launching a global trial of a possible treatment. Their first candidate—an antiviral medication known as remdesivir—was given to human subjects barely a month after the first cases were reported. But the standard and time-tested approach to measuring the worth of remdesivir and any other treatments for COVID-19 seemed glaringly at odds with the need to operate at the speed of a global catastrophe.
For 50 years, the gold standard for evaluating new drugs has been the two-armed randomized controlled trial (RCT). The model is nothing if not straightforward: Participants are assigned to one of the trial arms, the first of which gets the experimental drug while the second is given a placebo or the standard existing treatment. Augmented over the years by safeguards for patients and other rules to keep the results fair and accurate, this process has been the road by which most current prescription drugs have arrived on the market. But setting up such a trial for each promising COVID-19 treatment might take years, and by assigning half of these ailing patients to a placebo or a less effective treatment, hundreds could be denied their best chance to get better.
For Clifford Lane, NIAID deputy director for clinical research and special projects, that model simply wasn’t a match for the moment, especially with the sheer number of new and existing treatments that U.S. teams would need to explore simultaneously. “There were a lot of things we thought might be worth testing,” says Lane, who was selected to join a World Health Organization team on a trip to Wuhan to help study remdesivir. “Rather than running a whole series of independent trials, it made sense to use one design where you could put the most promising drugs first, bring in others later and get rid of those that weren’t working sooner rather than later.”
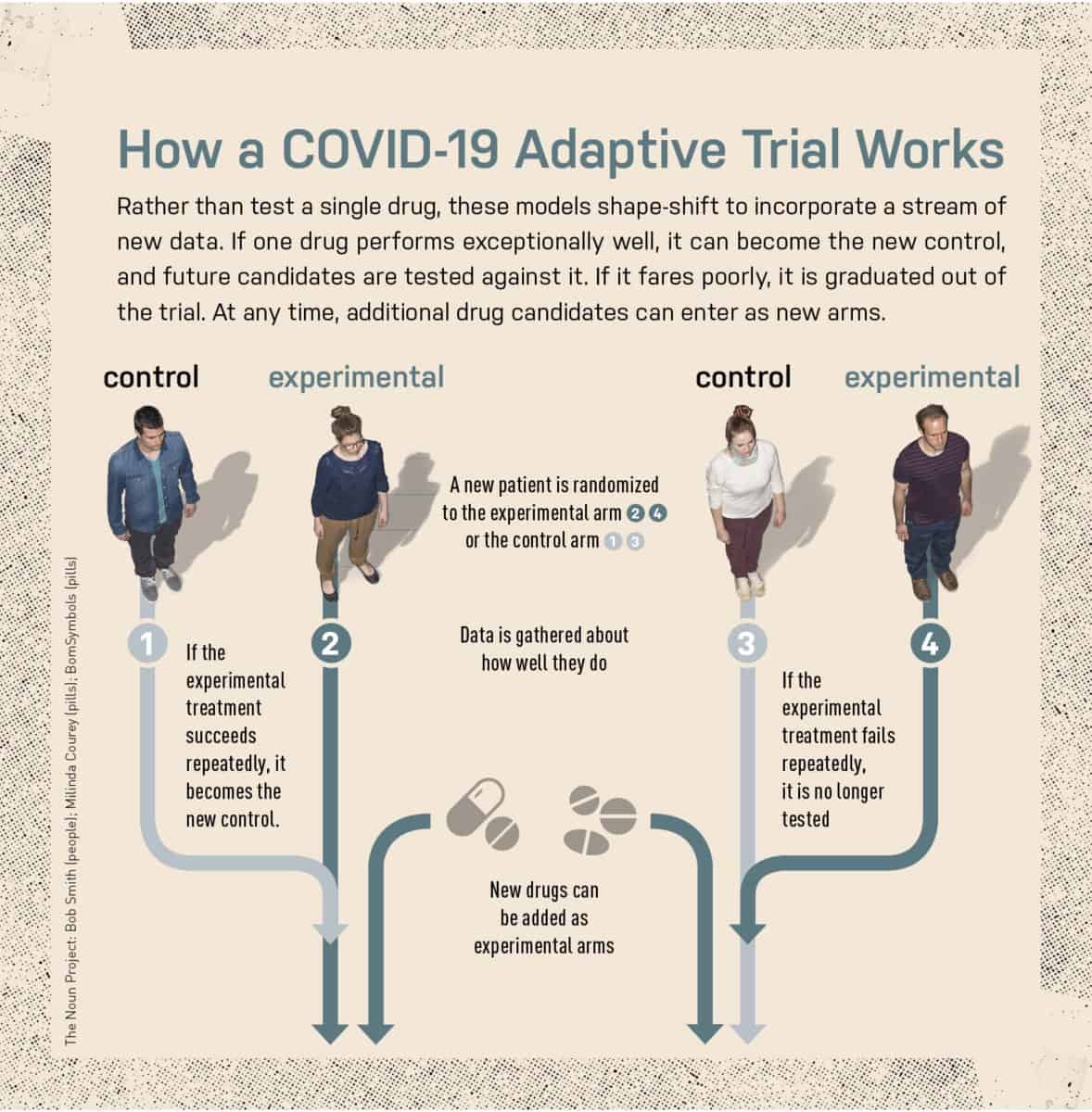
That alternative approach is known as an adaptive trial, an emerging model that adds (or removes) additional arms and protocols to a trial and evaluates the drugs’ effects by means of complex statistics. Something similar had worked well during the 2014 outbreak of Ebola in West Africa that caused more than 11,000 deaths. To search for effective therapies, Lane and his colleagues first gave trial participants ZMapp, an antibody-based drug, and compared their progress with those of a control group receiving “supportive care,” which included fever reduction techniques and intravenous hydration. When ZMapp proved to be no worse than the standard approach, the same study simply changed its shape during its next implementation in the recent Ebola outbreak in the Democratic Republic of Congo: The control group was switched over to ZMapp, which researchers compared against three additional drugs, including remdesivir. Ultimately this way of conducting a clinical trial rapidly proved that two other antibody medications, MAb114 and REGN-EB3, performed best. As soon as that was known, all subsequent patients were randomized to receive MAb114 or REGN-EB3.
That dramatic success demonstrated that nontraditional trial designs could be critically useful during an infectious disease crisis. “Despite being in the middle of an outbreak, and amid tremendous social disruption, it was possible to conduct rigorous research and come up with valid answers,” Lane says. Now, NIAID scientists have engineered similar trial designs to test COVID-19 treatments. One approach—to use the same control group to gauge the effectiveness of several candidate treatments—enables researchers to enroll fewer trial participants overall. The adaptive trial design also means that drugs that work well early on could become the standard of care for later patients.
This is just one way to configure an adaptive trial, and the ingenious products of this experimental field are known, collectively, as complex, innovative designs (CIDs). While a fleet of CIDs have been enlisted in the pandemic, they owe a debt to 10 years of groundwork and refinement in other fields.
The largest, longest-running adaptive platform trial, I-SPY2, has enrolled more than 1,700 women with high-risk breast cancer and evaluated 20 therapies since 2010. Regulatory agencies have historically been skeptical of the many innovations used in I-SPY2, but that has been changing, and in September 2019, the Food and Drug Administration issued its draft guidelines to help CID trials get past regulatory hurdles. Efforts inspired by the I-SPY2 approach are now being used in prostate cancer, pancreatic cancer, Alzheimer’s disease and glioblastoma. This spring the first patients were scheduled to enroll in the HEALEY ALS Platform Trial, the first CID platform for ALS, led by Merit Cudkowicz, chief of neurology and director of the Sean M. Healy & AMG Center for ALS at Massachusetts General Hospital.
These new types of trials are also now being used to evaluate treatments for COVID-19. With millions of lives in the balance, the world is watching to see just how well they work.

Illustration by Kacper Kiec
IN THE HISTORY OF clinical trials, one of the first landmark experiments commenced aboard the Salisbury, a 50-gun British warship, on May 20, 1747—and is the reason International Clinical Trials Day is celebrated on that day. The ship’s surgeon, James Lind, wanted to resolve a debate about the best way to combat scurvy, an incapacitating and sometimes deadly disease. He gave oranges and lemons to a handful of sailors and different treatments to others, then saw who fared better in the end. (The citrus diet won out.)
Two centuries later, Austin Bradford Hill, an English statistician and epidemiologist, introduced another crucial idea: The treating physician shouldn’t choose or know which patients received the treatment. The random, secret assignment of test subjects to one or the other arm of a trial would reduce the chance of bias by a doctor, who might otherwise skew results by giving a promising treatment to patients considered most likely to improve. Hill used the method in 1948 to gauge the effects of the antibiotic streptomycin on tuberculosis. Additional rules further improved objectivity. Researchers had to keep the questions they asked patients relatively simple, for better standardization, and weren’t allowed to change anything once a trial was under way. All of these led to a process through which the potential benefits of a drug could be observed in the most rigorous fashion.
But that rigor increasingly comes with an administrative burden and a steep price. Today the road to FDA approval of a new treatment normally proceeds in three (sometimes four) phases, which progressively ramp up requirements for showing safety and efficacy. Most drugs don’t make it to phase 3, which enrolls hundreds or thousands of patients and measures results against existing treatments. All told, only about 12% of medicines that enter clinical trials come out the other end with the FDA’s stamp of approval, according to a 2016 study published in the Journal of Health Economics, which also pegged the average cost of bringing a new drug to market at $2.6 billion (although other research has argued that figure is probably lower).
A portion of that expense can be attributed to conducting clinical trials, with the average cost of a phase 3 trial pegged at $19 million by a 2018 JAMA Internal Medicine study, the first to compile such an estimate. Tim Cloughesy, a neuro-oncologist at the University of California, Los Angeles, and global principal investigator of the GBM AGILE Trial for glioblastoma, says that six recent phase 3 trials for that deadly brain cancer collectively added up to $600 million. Such price tags, and the need to recruit large numbers of patients, make modern RCTs untenable in many cases, Cloughesy says. “We need a better way to evaluate therapeutics,” he says.
But high costs and long timelines aren’t the only drawbacks of traditional RCTs. There is often an inherent conflict between the scientific needs of drug trials and participants’ ongoing clinical care, a disparity that has been cited in multiple studies as a barrier that discourages physicians from enrolling their patients. That reluctance means research subjects can be very hard to find. A 2015 report published in the Journal of the National Cancer Institute found that one in five oncology trials fails because it can’t recruit enough participants.
“Our objective has always been to deliver better therapy to patients—those in the trial and those outside the trial”
Donald Berry, a professor in the department of biostatistics at the University of Texas MD Anderson Cancer Center in Houston
PROPONENTS OF CID BELIEVE that it can address many of the drawbacks of the traditional drug development pathway by designing a single study that is made up of phase 1, 2 and 3 trials all “soldered together” into one. They contend that asking multiple questions and evaluating many therapies in a single trial requires fewer patients, reduces overall costs and increases the likelihood that trial participants will receive beneficial treatments.
“Our objective has always been to deliver better therapy to patients—those in the trial and those outside the trial,” says Donald Berry, a professor in the department of biostatistics at the University of Texas MD Anderson Cancer Center in Houston. Barry has helped develop alternative trial designs that use something known as Bayesian statistical analysis to calculate probabilities and refine models based on early results.
One trial to emerge from experiments with Bayesian analysis was I-SPY2, a collaboration between Berry and Laura Esserman, a surgeon and oncologist who directs the Carol Franc Buck Breast Care Center at the University of California, San Francisco, School of Medicine. Known as an adaptive platform trial, I-SPY2 started in 2010 by testing one potential breast cancer therapy—and another 19 since then—in women whose tumors had a high chance of metastasizing. The adaptive design included randomization algorithms that adjusted the likelihood that a patient would be randomized to a particular arm of the trial based on how well that arm was performing. Therapies that performed well in phase 2 of I-SPY2 would then “graduate” to phase 3 trials outside of I-SPY2.
Berry and Esserman also designed I-SPY2 so that the control arm could be changed as the trial progressed. Initially, controls received the best treatment currently available. But in this trial, if a new drug proved better, it became the new standard of care and was substituted for the original control group therapy. That aspect became critical for infectious disease trials, including those for COVID-19. It ensures two things—that participants in the control group wouldn’t get what had become substandard treatment, and that additional drug candidates would be compared with the new state of the art. “We found that patients’ outcomes were getting better and better over the course of the trial,” Berry says.
The I-SPY2 team published their answer for a number of therapies in The New England Journal of Medicine in July 2016. They showed that the new therapies had met prespecified criteria for advancing to phase 3 trials. An accompanying editorial described I-SPY2 as “a promising adaptive strategy for matching targeted therapies for breast cancer with the patients most likely to benefit from them.”
THE ENCOURAGING RESULTS OF I-SPY2 threw open the doors for adaptive platform clinical trials in many other diseases, including COVID-19. “There has been some reluctance to embrace new ideas, but that has been changing,” says Patrick Phillips, a tuberculosis researcher at UCSF who helped run an adaptive phase 2 trial sponsored by PanACEA, a pan-African effort to find a better antibiotic treatment for TB.
To create a rulebook, the FDA weighed in with interim guidelines in September 2019 about how such trials might be used. The FDA recommends that sponsors bring the agency into the loop at the earliest stages of trial design and provide detailed explanations of the criteria that will be used to trigger critical changes—such as adjusting the probabilities that someone will be randomized into one arm or another, and when a drug will be added to or removed from a trial.
Yet even as more researchers consider the possibilities of CID trials, inherent limitations may restrict their use for now. For example, the Bayesian statistical analyses that are essential to CID trials require specific training and advanced computing skills, and their complexity may create a “black box” effect that prevents the clinicians conducting trials from truly gauging the results. As a byproduct of these algorithm-driven treatment courses, a drug that appears to be effective in one CID trial may fail in another, says John Ioannidis, professor of medicine and a clinical trials expert at Stanford University, who considers this lack of reproducibility a major drawback. Sarah Blagden, an oncologist at the University of Oxford, says she believes CID trials hold great potential, but cautions that too few have been done for scientists to verify that, in fact, they are better, faster and cheaper than the traditional trial pathway. Getting to that point will require more time—and more trials. “Although cost-effectiveness assessments are being done, we do not yet have a definitive answer as to whether conducting a single, complex study is better than the traditional, separate study route,” she says. “But instinctively, it seems likely.”
Those new trials, however, are beginning to materialize. The COVID-19 global clinical trial will be a major testing ground. Another effort slated to take off this year is a large platform trial at the Healey Center for ALS at MGH. A team led by Cudkowicz will initially evaluate three new drugs, with several more slated to be added later. Each potential ALS therapy will have its own arm, with 120 participants taking the new drug and 40 serving as a control group. But those same 40 controls will be utilized in all three arms, and together those arms of the trial will require just 480 participants—compared with a total of 720 that would otherwise have been needed. “We wanted to show that you could have a very patient-friendly design and get clear answers faster,” Cudkowicz says. In addition, adding new treatments can be done much faster because a new drug protocol is just added as an amendment to the master protocol.
The groundwork that all of this lays for the COVID-19 response could not be more critical. “In these trial designs we have a tool that, we hope, can not only give us good information but also let us help as many of our enrollees as we can,” NIAID’s Lane says. That proposition, if it holds out, could be a major step forward for other research and treatment questions long after the current crisis is over.
Dossier
“Challenges with Novel Clinical Trial Designs: Master Protocols,” by Michael Cecchini et al., Clinical Cancer Research, January 2019. This article lays out some of the common issues relating to innovative clinical trials.
“Effective Delivery of Complex Innovative Design (CID) Cancer Trials—A Consensus Statement,” by Sarah P. Blagden et al., British Journal of Cancer, January 2020. The authors provide an overview of complex, innovative design.
“I-SPY 2—Toward More Rapid Progress in Breast Cancer Treatment,” by Lisa A. Carey and Eric P. Winer, The New England Journal of Medicine, July 2016. This editorial looks at how the I-SPY 2 trial is advancing breast cancer treatment specifically and medical science as a whole.