Published On January 31, 2017
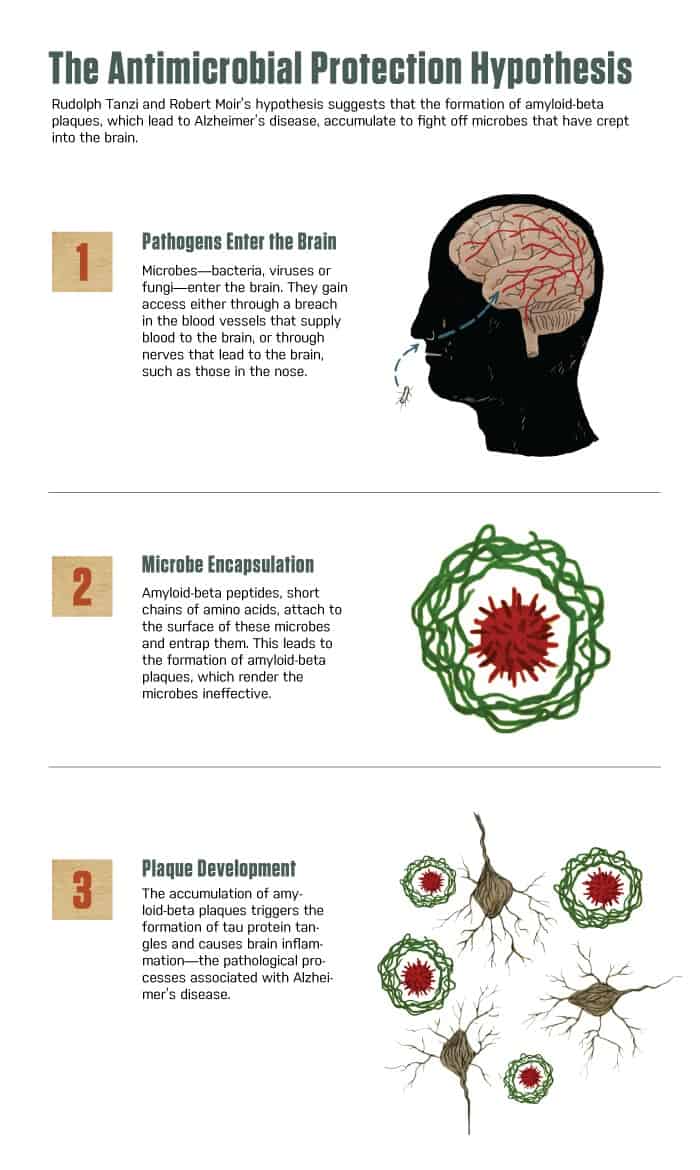
TO THOSE OUTSIDE THE FIELD OF NEUROSCIENCE, the process may have seemed a little ghoulish. Rudolph Tanzi and Robert Moir took autopsied brain tissue from patients who had died of Alzheimer’s disease and “homogenized” it, grinding up the tissue using a sterile, laboratory-grade mortar and pestle. “Not terribly elegant, but highly effective,” says Moir, assistant professor in neurology at Massachusetts General Hospital. The tissue came from a region of the brain that was ravaged by Alzheimer’s. In the form of a “broth,” the homogenized tissue was lethal to microbes—as much as 100 times more potent than penicillin, says Moir.
The tissue apparently owed its antimicrobial prowess to amyloid-beta, a protein whose plaques clog the brains of people with Alzheimer’s. That broth far outperformed others created from the brains of people who had shown no signs of dementia when they died, or even from regions of the diseased brains that didn’t have the plaques. For Moir and Tanzi, who is the director of the Genetics and Aging Research Unit at MGH, the results were a critical step in advancing a notion about the causes of Alzheimer’s.
The conventional view regards amyloid-beta as purposeless junk that slowly accumulates in the brain as humans age. That buildup triggers the processes that eventually lead to Alzheimer’s in some people. The “broth study,” published in 2010, offered an alternative explanation—that the amyloid-beta plaques might form to help the brain fight infection. And in a new paper, Tanzi, Moir and their colleagues argue that amyloid-beta may indeed play a crucial role in protecting the brain from attacks by bacteria, yeast, fungi and viruses.
Their latest work supports a tantalizing new way to think about this disease. If amyloid-beta indeed accumulates to fight a low-grade, persistent infection, it might lead to novel therapies for treating or even preventing Alzheimer’s.
THE FIRST CLINICAL DESCRIPTION of Alzheimer’s disease—an irreversible, neurodegenerative disorder—came in 1906, from a German psychiatrist and neuropathologist Alois Alzheimer. He had examined the brain of a woman with serious dementia when she died and described two distinct abnormalities, now recognized as the plaques and tangles that are characteristic of the disease.
The nature of those plaques finally came into focus in 1984, when George Glenner, a research scientist at the University of California, San Diego, identified the peptide called amyloid-beta and hypothesized that Alzheimer’s was caused by “amyloidosis” of the brain, a process in which insoluble forms of an amyloid protein accumulate. Then, in 1987, Tanzi and his colleagues published their discovery of the first Alzheimer’s-associated gene, which leads to the formation of amyloid-beta precursor protein, or APP. When APP breaks into smaller peptides, one byproduct is amyloid-beta.
Glenner’s research eventually morphed into the “amyloid cascade hypothesis,” which says that the formation of amyloid-beta plaques leads to tangled forms of another protein, tau, and ultimately to inflammation in the brain.
In Glenner’s scenario, amyloid-beta is the primary culprit, and that notion has been strengthened by studies of early-onset Alzheimer’s, which accounts for about 5% of Alzheimer’s cases. This form of the disease runs in families and has been linked to genetic mutations that help increase deposits of amyloid-beta in the brain. In addition to the APP gene, another variant, called APOE-4, has also been implicated in the late-onset form of the disease. It appears to keep the brain from clearing the amyloid-beta deposits that accumulate as people age. Not everyone with APOE-4 develops Alzheimer’s, however, and not all people who get the disease carry APOE-4.
But some researchers had remained unconvinced that amyloid-beta deposits initiate Alzheimer’s. In mouse models of the disease, tau tangles didn’t form after the plaques appeared. And no one had shown a clear link between amyloid-beta plaques and tau tangles, even when using human brain cells grown in a petri dish.
In 2014, however, Tanzi, his colleague Doo Yeon Kim and their team published a paper based on their work with an ingenious three-dimensional gel matrix designed to culture a set of human brain cells. The cells had been genetically engineered to produce large amounts of APP, and also carried mutations linked to familial Alzheimer’s. In this “Alzheimer’s in a dish,” amyloid-beta plaques indeed formed first, followed by tau tangles. When the scientists kept amyloid-beta from forming, there were no tangles. For Tanzi, this proved the amyloid cascade hypothesis. But it still left the question of why the brain makes amyloid in the first place.
IT TURNS OUT THAT the sequence of amino acids that makes up the amyloid-beta protein isn’t unique to humans. It’s 400 million years old, and is shared among 70% of all vertebrate species. And that sequence can form long chains called oligomers as well as fibers called fibrils. “That sequence being conserved across that length of time suggests very strongly that it’s doing something important,” says Robert Moir. He argues that if those oligomers and fibrils weren’t needed, evolution would have eliminated them long ago. “But I had no idea what their function might be,” he says.
Then, several years ago, some idle web surfing led to a possible answer. On Friday afternoons, Moir indulges in what he calls “PubMed play hour.” The PubMed website contains links to tens of millions of journal articles, books and other scientific literature. “I follow whatever links take my fancy,” Moir says. One Friday, it took him to a substance called LL-37, a known antimicrobial found in the brain. Though not identical to amyloid-beta, LL-37 behaved in very similar ways.
Did amyloid-beta serve the same purpose? By 2008, when Moir stumbled on the parallels between amyloid-beta and LL-37, Tanzi had discovered additional genes associated with Alzheimer’s that were also related to innate immunity, the part of the human immune system that is shared with worms, flies, spiders and other primitive creatures. Innate immunity wards off microbial intruders.
Moir and Tanzi decided to explore the connection between innate immunity and Alzheimer’s disease. It was then that they came up with their brain broth experiment and discovered that tissue from diseased regions of Alzheimer’s patients’ brains could kill microbes with stunning potency.
That was reported in 2010. Additional experiments further tested their hypothesis that amyloid-beta is a “foot soldier” of innate immunity, as Moir puts it. He and his colleagues showed that nerve cells engineered to produce amyloid-beta were better able than other cells to stand up to the infectious Candida albicans, a fungus. The amyloid-beta bound itself to C. albicans and grew fibrils that entombed the fungal invader—and as part of that process, created plaques of amyloid-beta.
The team saw similar behavior in animals. Those whose brains could produce amyloid-beta showed an advantage in resisting infections. Results were particularly striking in mice engineered to develop Alzheimer’s disease at about five months of age. Tanzi and Moir worked with youthful, four-week-old mice, which were a long way from developing amyloid-beta plaques. But when researchers injected S. typhimurium, a strain of salmonella, into the brains of these mice, they developed plaques of amyloid-beta overnight, presumably to protect against the salmonella. Sometimes, a plaque would form around a single bacterium.
All of this promoted the idea that amyloid-beta plaques weren’t waste products in the brain, but rather were produced by an active immune defense system. Yet medical journals were cool to the paper Tanzi and Moir prepared to present their hypothesis. “It was rejected six times without review,” says Moir. Even Science Translational Medicine, which finally published the paper, turned it down the first time around. But after the journal’s editor heard the researchers discuss their work at a conference, the journal invited them to submit the paper again. It appeared in May 2016.
TANZI AND MOIR HAD no idea how their hypothesis would be received. “We were fully expecting a variation of the Schopenhauer rule, where a new discovery is first ridiculed, and then violently opposed, before it’s accepted as self-evident,” says Tanzi.
Instead, an essay in Nature Reviews Neurology called the study “balanced and nuanced.” The author, Todd Golde of the University of Florida in Gainesville, says that he found the base observations provocative, and believes that “they deserve to be followed up right now.”
Brian Balin, director of the Center for Chronic Disorders of Aging at the Philadelphia College of Osteopathic Medicine, wasn’t surprised by the paper’s “infection hypothesis.” Balin’s own work had already suggested a link between microbes and Alzheimer’s disease, as had the work of Ruth Itzhaki at the University of Manchester in the United Kingdom and a few others. As early as 1991, Itzhaki’s group had shown that the DNA sequence of the herpes simplex virus type 1 is often present in brains of people who have died of Alzheimer’s. Balin’s group had implicated the Chlamydia pneumoniae bacterium, an airborne infectious microbe.
Balin’s team has also shown that if mice breathe in C. pneumoniae and become infected, their brains begin to generate amyloid-beta. Both the chlamydia and herpes microbes can enter brains through the sense of smell, says Balin, and then travel up olfactory nerves to a part of the brain called the entorhinal cortex—which happens to be one of the earliest to sustain damage in Alzheimer’s disease.
Other bugs, however, don’t have such ready access to the brain. They’re carried in the blood and must get past the so-called blood-brain barrier, composed of specialized cells lining the walls of capillaries in the brain. Normally, this creates a formidable defense, but it’s not impermeable. In a study of early-stage Alzheimer’s, Jacobus Jansen of the Maastricht University Medical Center in the Netherlands and his colleagues studied people aged 59 to 85 who were in early stages of Alzheimer’s disease alongside a control group of people who didn’t have Alzheimer’s symptoms. Researchers injected a tracer called gadolinium intravenously into the subjects and then scanned their brains to see how much got past the blood-brain barrier—which was noticeably leakier in those who had Alzheimer’s.
Jansen is among those who think that a compromised network of blood vessels in the brain triggers Alzheimer’s by allowing toxins to cross the blood-brain barrier, potentially leading to damage to neurons and inflammation. According to this hypothesis, that also prevents the brain from effectively clearing amyloid-beta, leading to tau tangles and more inflammation. Previous studies have shown there indeed is a correlation between deteriorating blood vessels in the brain and cognitive decline, and the new work by Jansen and his colleagues finds that the leakier the blood-brain barrier becomes, the worse the cognitive problems may be. In this vascular-decline hypothesis, the amyloid cascade is thought to be a consequence of the damaged blood vessels and only part of the process contributing to Alzheimer’s.
But Jansen believes the new work from Tanzi and Moir may help connect these two views. Perhaps the blood vessels begin to deteriorate first, causing leaks in the blood-brain barrier that allow microbes to get in. Amyloid-beta then forms plaques to fight off the invaders, and that triggers the rest of the disease process. “This hypothesis is a nice combination,” says Jansen. “It provides an explanation for why both processes occur at the same time.”
Nevertheless, the idea that infections cause Alzheimer’s disease remains a minority view, says Florida’s Todd Golde, who notes that much of the data linking pathogens to Alzheimer’s disease may be flawed. Many people with Alzheimer’s die of sepsis, which can be caused by infections, and he believes brain tissue samples could have been contaminated during autopsies with microbes from elsewhere in the body.
Golde also wonders whether “the artificial sort of systems” used by Tanzi, Moir and their team—such as their cultured brain cells that produced the amyloid-beta plaques—are reliable proxies. Golde notes that while normal concentrations of amyloid-beta in human brains can aggregate to form plaques, that doesn’t happen in the lab without help. “In order to make it aggregate in a test tube, we need thousand-fold higher levels,” he says.
Perhaps there is something in human brains that triggers the plaques to form, and Tanzi and Moir believe they have shown what that is—infection. But there are other possible explanations, including the activity of apolipoprotein E (APOE), a substance known to regulate amyloid deposits.
AS MANY DIFFERENT, AND sometimes conflicting, strands of research continue to probe how and why plaques and tangles form in the brain, the ultimate goal is to find a way to keep that from happening. So far, although there are treatments that may ease some symptoms of Alzheimer’s disease, nothing comes close to a cure.
But if additional studies confirm amyloid-beta’s antimicrobial function—and the role of infections in causing amyloid-beta plaques to form—this model might open up new ways of thinking about Alzheimer’s therapies. For example, if particular microbes are implicated, perhaps they could be attacked with brain-permeable antibiotics before plaque formation gets too far along. Or patients could be injected with antibodies to clear up the infections. Vaccinations designed for specific microbes might head off trouble altogether. But Balin says that attempts to create vaccines against strains of herpes and chlamydia found in the brains of Alzheimer’s patients have proven difficult, and there’s nothing currently on the horizon.
Moreover, Tanzi cautions against building therapies around a hypothesis that has yet to be confirmed in people. The next step, he says, will be to use his team’s three-dimensional “Alzheimer’s in a dish” model to see whether microbes can induce amyloid-beta plaques to form in human brain tissue, and then whether those plaques lead to tau tangles and inflammation. The researchers also hope to use gene sequencing technology to compare the brains of those who have died of Alzheimer’s with those of people who died without neurological diseases, in the hope of finding DNA sequences of particular microbes in the diseased brains.
“Once we see what bugs or viruses or yeast might be living in the brain, we can ask whether they indeed induce amyloid to form,” says Tanzi. “We certainly don’t want people to start taking antibiotics or trying to address infection as a way to stop the disease. We are not there yet.”
And even if this hypothesis is confirmed, it’s clear there are also other reasons, including overactive synapses between brain cells, that can lead to the accumulation of amyloid-beta plaques. In addition, there are questions about why some brains are better than others at controlling amyloid-beta plaques, and why some people with plaques don’t develop Alzheimer’s. Even if infections are a primary cause of the plaques, it appears that only some of those who are infected with herpes, chlamydia, spirochetes or yeast may be vulnerable.
With so much still unknown, Tanzi recalls advice he got from his doctoral thesis advisor, MGH geneticist James Gusella, nearly 30 years ago: “Date your hypothesis, don’t marry it.” It should be fascinating to see how this relationship develops.
Dossier
“Alzheimer Disease: Host Immune Defence, Amyloid-β Peptide and Alzheimer Disease,” by Todd E. Golde, Nature Reviews Neurology, July 2016. A review of evidence that amyloid-beta plays a physiological role in the brain.
“Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease,” by Harm J. van de Haar et al., Radiology, March 2016. A compromised blood-brain barrier, which prevents some materials in the blood from entering the brain, is associated with cognitive decline.
“Amyloid-β Peptide Protects Against Microbial Infection in Mouse and Worm Models of Alzheimer’s Disease,” by Deepak K.V. Kumar et al., Science Translational Medicine, May 2016. Amyloid-beta may accumulate to help the brain fight infection.